Chloe siempre lleva un cuaderno en la mano. A sus catorce años es escritora. Acaba de terminar su primer libro, una novela que ahora está en edición. Se trata de un drama adolescente —del género que en inglés se conoce como coming-of-age—, la publicación llegó por un camino inesperado.
Un pulmonólogo que trataba a Chloe contactó con la organización sin ánimo de lucro Make-A-Wish, dedicada a cumplir deseos a niños con enfermedades graves. Cuando le preguntaron a ella qué quería, Chloe pidió algo insólito: ceder su deseo a otros niños con problemas de salud mental. Pero su doctor intercedió: «¿Por qué no pides que te publiquen el libro que estás escribiendo?».
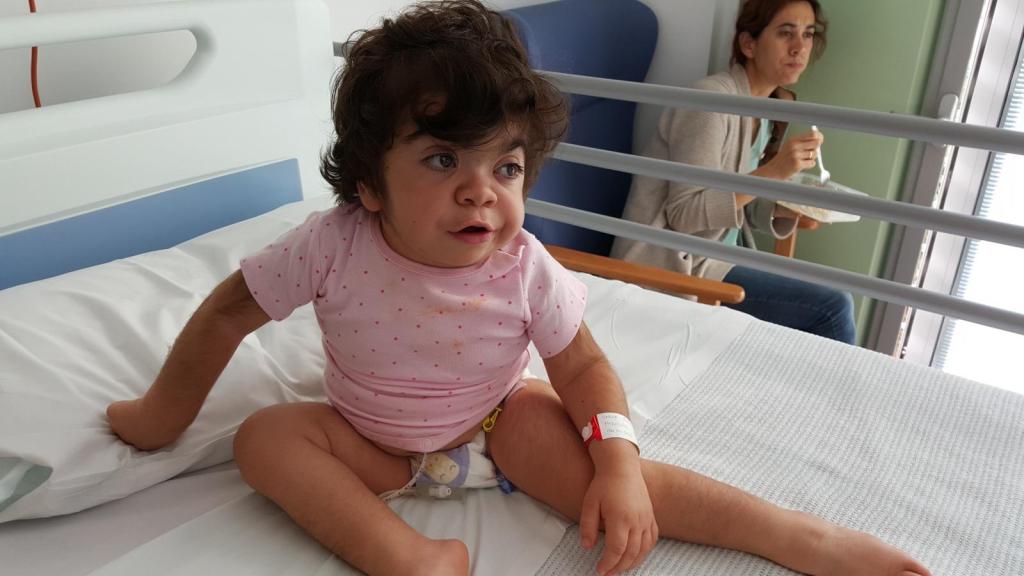
Chloe es la excepción que confirma la regla. A los nueve meses de nacer le diagnosticaron síndrome de Hurler. Uno de cada cuatro. Esa era la probabilidad. Cuando Teresa Álvarez (43) y Mariano Blanc (48) decidieron tener hijos, desconocían que ambos portaban una herencia silenciosa en los genes. Se conocieron en Madrid, trabajando en cine. «Ella llegó a mi mesa, empezó a trabajar conmigo. Y así coincidimos», comenta Mariano. Comenzaron a salir y a los dos meses le pidió que se casara con él. Lo retrasaron un año y el 7 de febrero de 2008 contrajeron matrimonio en Toledo. Después llegaron Carlos, nacido en Nueva Zelanda en 2009, y Chloe, en 2011 en Madrid.
El gen que la pareja porta se trata de una copia defectuosa del gen de la α-L-iduronidasa que, por separado, no significa nada. Pero cuando el azar decide unirlas, hace que el cuerpo del niño pierda la capacidad de limpiarse y los residuos se acumulen. En el corazón. En los huesos. En los ojos. En el cerebro.
Su incidencia ronda un caso por cada 100.000 nacimientos. En España, se estima que unas 250 personas conviven con la forma más grave de esta condición. No tiene cura, pero existen dos vías de tratamiento: el trasplante de médula ósea, realizado antes de los dos años y medio, y la terapia de reemplazo enzimático.
«Al principio nos dijeron que tenía algo en el corazón, era un poco diferente pero funcionaba bien. La dejaron en observación. No fue un indicio de que fuera Hurler, sino que era algo cardíaco menor», recuerda Teresa. Su voz suena serena, pero hay pausas que delatan el recuerdo de la incertidumbre. «Luego, a los nueve meses, empezó a enfermarse constantemente. Se desarrollaba despacio y hubo varios médicos que empezaron a preguntar a quién se parecía. Fue gracias a una cardióloga en Inglaterra, que reconoció los rasgos de Hurler», añade.
Los rasgos. Ese intento de asemejar y comparar lo que no tiene diagnóstico. Los niños con síndrome de Hurler suelen tener cabeza grande, macrocefalia, y sus facciones se vuelven toscas con el tiempo. La lengua, grande, a veces sobresale. Los labios se engrosan. Los ojos, velados por una opacidad corneal progresiva, pierden brillo. El cuerpo también se deforma: tronco corto, baja estatura, vientre abultado por el agrandamiento del hígado y el bazo. Antes de que todo eso sea evidente, queda la mirada de una madre que intuye que algo no va bien.
«Chloe era como una muñeca de trapo, no se sostenía. ¿Sabes lo típico? Los niños a los dos meses sostienen la cabeza. A ella le costaba todo. Y luego, con la comida: no tragaba sólidos. Yo iba al médico, a la enfermera, y me decían: ‘todos los bebés se desarrollan a su tiempo, no te preocupes’. Con lo de comer, una enfermera me soltó que era porque ella no quería. Luego resultó que no podía. Ahora lo pienso y me da una rabia… A veces tenía la sensación de que me trataban como a una madre histérica«, confiesa.
Esa sensación de no sentirse escuchada es un sentimiento común en los familiares de niños con enfermedades raras. El doctor Cristóbal Colón, de la Unidad de Diagnóstico y Tratamiento de las Enfermedades Metabólicas Congénitas del Hospital Clínico Universitario de Santiago de Compostela, lo confirma: «Primero, reciben un golpe de que ese niño no va bien, le han hecho todas las pruebas y no hay nada. Entonces, se encuentran desesperados. A veces hay padres que lo único que buscan es, por lo menos, tener un nombre y apellidos de qué le pasa al niño».
Colón y su equipo llevan años peleando por adelantarse al sufrimiento. «Hicimos campañas en todos los congresos de pediatría para que prestaran alerta en su consulta a ciertos signos o síntomas característicos. Pusimos en marcha el proyecto FIND: tú tienes un niño problemático en tu consulta que no sabes lo que tiene y de forma gratuita nos mandas una orina y sangre impregnadas en papel y nosotros lo analizamos. Si una enzima está baja, le decimos: ‘oye, mira, este niño tiene un posible Hurler’». El resultado ha sido efectivo: «Hemos logrado detectar casos en toda España. Antes, la media de edad en el momento del diagnóstico andaba en la adolescencia y en los últimos años todos tenían menos de un año. El pronóstico de la enfermedad cambió drásticamente. Nada más empezar el año pasado el cribado, ya apareció un caso que se trasplantó y el niño está muy bien».
ESPAÑA, UN REFERENTE EN ENFERMEDADES METABÓLICAS
España cuenta con centros de referencia de primer nivel internacional en el diagnóstico y tratamiento de las mucopolisacaridosis. Cada año se diagnostican una media de 80 nuevos pacientes con errores congénitos del metabolismo, un tercio de los cuales son lactantes. Sus laboratorios son uno de los tres centros en Europa que realizan estudios de aminoácidos y neurotransmisores, y trabajan con paneles génicos específicos para confirmar genéticamente cada patología.
Allí, la doctora Mercè Pineda, neuropediatra e investigadora, ha liderado estudios pioneros en terapia génica para el síndrome de Sanfilippo. Aunque la terapia génica para Hurler aún está en fases experimentales, la investigación avanza. En Madrid, el Hospital Universitario La Paz ha dado un paso adelante con la creación de la Unidad Multidisciplinar Infantil de Enfermedades Minoritarias, la primera de estas características en España. Está pensada para atender a unos 350 niños con enfermedades genéticas complejas, coordinando hasta trece servicios diferentes, desde Reumatología hasta Oftalmología pediátrica, pasando por Genética y Farmacia. El Centro de Diagnóstico y Estudios Metabólicos Avanzados de Cantabria, dirigido por Domingo González Lamuño, que junto al Hospital Germans Trias i Pujol de Badalona —donde el doctor Guillem Pintos ha coordinado programas de acceso precoz a tratamientos para MPS IV— completa un mapa de excelencia que sitúa a España en la vanguardia de la investigación en enfermedades metabólicas raras.
«Somos pocos centros, nos conocemos bien, intercambiamos mucha información», explica Colón. «Por desgracia, hay mucho todavía que avanzar porque, cuanto más conoces, más enfermedades te aparecen. A ver si ,más adelante la inteligencia artificial se acelerará todo el proceso. Tú cuando pides un genoma completo, tardas meses en hacer un informe. Y a veces cuando llegas con él, el niño ya se ha muerto», agrega el doctor.
LA VIDA DESPUÉS DEL TRASPLANTE
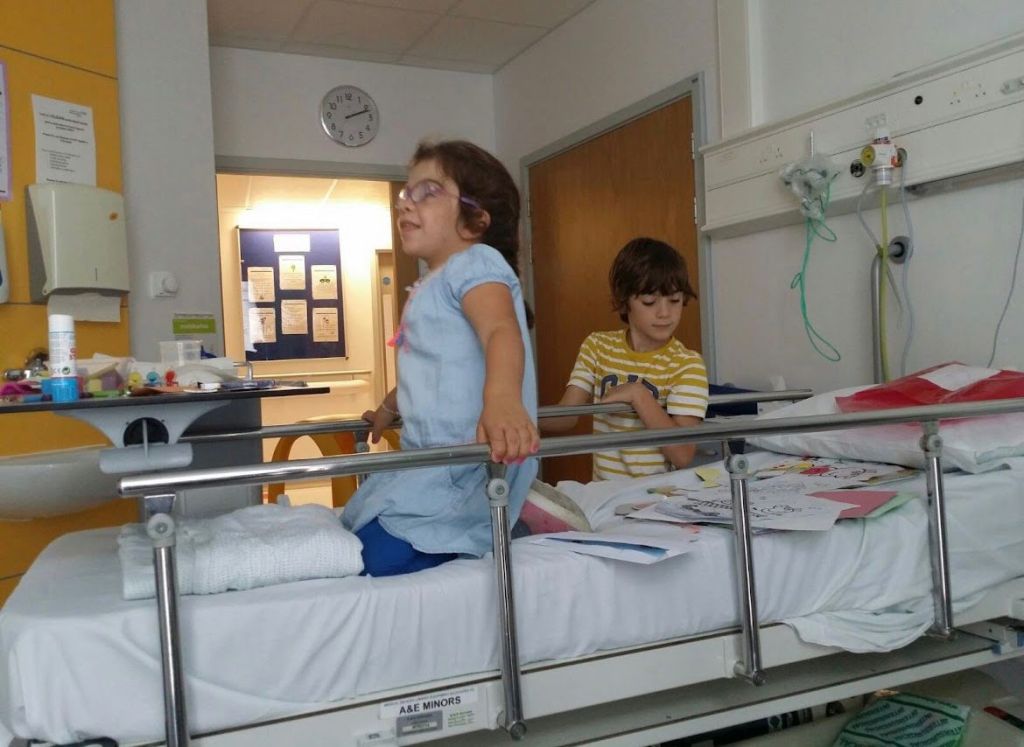
Chloe recibió el trasplante de médula en 2012. Pasó casi un año entero ingresada y otro más para recuperarse. Mariano lo recuerda con una mezcla de gratitud y asombro: «Hemos conocido gente que no entiendes cómo dan tanto. Y no solo por dinero, hay gente muy vocacional, muy buena. Les tiene que hacer mella ver tantos derrotas de niños, pero ellos siguen ahí». En su memoria, cuando todo se desdibuja, resuena el nombre de Raquel, una enfermera que estuvo con ellos durante el trasplante en Oxford, y el del doctor Kissatt, que lideraba el equipo.
«Económicamente es la pérdida de un sueldo en una familia», dice Teresa. «Son cosas que no puedes cuantificar: estar en el hospital dos días enteros a la semana, aunque ella estuviera bien, por pruebas, por consultas. No puedes dejar a un canguro en el hospital con los médicos. Hasta que Chloe tuvo diez años o así, para mí era imposible volver al trabajo».

El trasplante no es el final del camino, sino el principio de una nueva etapa. Requiere un seguimiento multidisciplinar constante: cardiología, oftalmología, traumatología, rehabilitación. Eso implica una logística que absorbe a la familia. Los padres se convierten en gestores de salud a tiempo completo. «Hay que recordarle la medicación, la fisioterapia, todo. Si piensas en un niño de 14 años, es prácticamente independiente. Con ella tienes que estar muy involucrado. Cosas que otros niños aprenden de forma orgánica, a ella se las tienes que enseñar como si le estuvieras dando una clase», explica su madre.
«Hace unos días, Chloe quiso ir sola a la biblioteca. Era la primera vez. Llegó a la esquina y se paró. No supo medir si los coches venían rápido. No sabía si aparecerían de repente. No veía bien el ángulo. Tuvo miedo y volvió al colegio», cuenta Teresa.
«Eso está bien, porque le hemos dicho que si no está segura, no lo haga», sonríe recordando la anécdota y añade, con ese punto de ternura que tienen las madres cuando miran el mundo con los ojos del hijo: «Nunca pensé que con 14 años tendría que enseñarle a cruzar la calle«.
«A veces, Chloe parece muy madura. Otras, es como si fuera una niña pequeña. Ha aprendido a sonreír y a disimular. Preguntas si ha entendido y dice que sí. Pero no se ha enterado de nada». Y en esa frase late una verdad incómoda: que los niños con enfermedades raras desarrollan estrategias de camuflaje, mecanismos para no preocupar, para encajar, para que no les pregunten demasiado.
Mariano añade: “Al principio quería cuidarla como en una cunita, que nada le duela, que no se exponga. Teresa tiene las ideas muy claras y me ha enseñado que lo mejor que hay que hacer con Chloe es vivir una vida normal y en un colegio absolutamente normal, rodeada de situaciones absolutamente normales, evitar el romance de la enfermedad alrededor. Sobre todo nunca edulcorar la enfermedad ni siquiera con ella”.
Chloe tiene un hermano, Carlos, de 18 años. Ha crecido con ella y, sin saberlo, se ha convertido en un chico distinto. «Tiene mucha más empatía que otros chicos de su edad. Mucha más paciencia. Si las cosas tardan, no se desespera», afirma Teresa. El padre agrega: “Carlos es lo mejor que le ha pasado a Chloe. Ha pasado la vida en hospitales desde pequeño, saltando de cama en cama, partiéndose de risa con su hermana”. En ocasiones les cuestiona: “¿dónde va a vivir Chloe? ¿Va a hacer amigos? ¿Le vais a dejar ir sola?” Preguntas que les hacen plantearse el futuro.

TRES PAÍSES, TRES SISTEMAS SANITARIOS
Teresa, Mariano, su hijo Carlos y la escritora Chloe han construido su hogar a ambos lados del mundo, viviendo en Nueva Zelanda, España, Inglaterra y Estados Unidos debido a las oportunidades laborales en el mundo del cine. Han sido cuatro países muy distintos, pero la familia asegura que hay algo que no ha cambiado en ninguno: el cariño y la buena atención que siempre ha recibido Chloe.
«En España y en Inglaterra es gratis. En Estados Unidos se paga un seguro muy caro, pero las listas de espera son mínimas«, compara la madre. «Cuando es urgente —una cardiopatía, un trasplante—, en todos lados funciona. Pero para cosas como la cadera, que duele pero no es urgente, en Inglaterra te dan largas. Puede ser año y medio de espera. Aquí, en San Francisco, lo hicieron enseguida. Dijeron: ¿para qué esperar si sabemos que hay que operar?».
También son evidentes las diferencias en el trato humano. «Aquí, en Estados Unidos, hablan con ella. Antes de una operación, alguien se sienta a su lado y le pide que explique con sus palabras lo que le van a hacer. Para asegurarse de que entiende y da su consentimiento. Conocemos al anestesista, al cirujano, a todo el equipo. Como padres, eso te da tranquilidad».

«Los resultados los tienes tú a la vez que el médico. Te los explican, pero tú también los ves. Y tienen muy en cuenta la autonomía del adolescente. Cuando Chloe empezó a entender, se lo explicaban más a ella que a mí. Eso me parece bien. Ella tiene 14 años, tiene derecho a decidir sobre su vida. En España, creo que aún falta eso. Los padres hablan, y los niños hacen lo que se les dice», comenta su madre.
El paso a la edad adulta es uno de los momentos más críticos. Hay que preparar al paciente y a la familia para que asuman el control de su enfermedad. En las consultas se trabaja que el adolescente conozca su medicación, sus síntomas, sepa cuándo pedir ayuda.
Cuando Chloe era pequeña, solía ir a reuniones con otras familias con niños con síndrome de Hurler. «Nos venía bien, sobre todo a los padres. Ella veía a otros chicos como ella y punto. Pero al llegar a Estados Unidos, algo cambió. Chloe no quiso entrar a una reunión. No quería verlos. No quería identificarse con ellos«, recuerda su madre.
«Estuvo yendo a un psicólogo para trabajar esa aceptación y al final dejamos de asistir a las reuniones. Creemos que es algo que tiene que procesar más adelante, cuando esté lista».
En la Asociación MPS España llevan años siendo el hombro donde apoyarse, el altavoz que reclama investigación y el puente entre familias y médicos. Su presidenta, Ana María Mendoza, describe el diagnóstico como un mazazo que llega para quedarse: un hijo con una enfermedad rara, degenerativa, sin cura. Pero después del shock inicial llega el día a día, un territorio hostil de terapias, adaptaciones y batallas administrativas. La investigación, aunque lenta, no se detiene. Hay esperanza.
Teresa y Mariano realizan una pausa larga cuando se les pregunta qué dirían a unos padres que acaban de recibir el mismo diagnóstico.
Teresa comenta: «Que estén tranquilos. Porque las cosas parecen peor desde fuera que cuando estás dentro. Que confíen en los médicos, pero que estén muy pendientes de que el equipo sabe lo que hace. Que lean mucho, que hablen con la asociación. Que se aseguren de que están teniendo el tratamiento adecuado. Y sobre todo, mucha positividad. Porque al final es una vida muy bonita. Hay muchos momentos de felicidad. Ojalá Chloe no estuviera enferma. Pero no lo cambiaría por nada del mundo».
Mariano, por su parte añade: «Si puedes llevarlo con humor, es lo único que puede llegar a salvarte el matrimonio y va a hacer que tu hijo esté con padres que quieran estar alrededor. He visto tantos matrimonios fracasados, tantos, tantos. No le puedes cargar con los dolores más fuertes a tu pareja. Esas descargas las tienes que hacer poco a poco. Tienes que pedir ayuda fuera, tener tiempo para relajarte, cuidarte».
Chloe sigue escribiendo. En los cuadernos que se acumulan en su mesa, en las salas de espera, en los recreos del colegio. A sus catorce años ha entendido que la literatura no es un escape, sino un oficio: el de convertir la experiencia en algo que merezca ser contado.

Deja un comentario